La théorie des collisions n'entre
pas dans le détail de l'interaction entre les molécules qui réagissent, ni dans
la manière dont l'énergie se répartie au sein de chaque molécule. Elle ne peut
donc pas rendre compte convenablement des différences de vitesses observées
pour des réactions différentes ni même pour différentes molécules qui participent
à une même classe de réactions. A ce propos, vous pouvez revoir éventuellement
la conclusion du précédent sous-chapitre sur la théorie
des collisions.
La théorie dite du "complexe activé",
développée principalement par H. Eyring et M. Polanyi dans les années 1930 se
place au départ dans le même cadre conceptuel que la théorie des collisions
mais entre dans le détail des interactions entre les espèces réagissantes pour
décrire la formation d'un complexe intermédiaire d'énergie élevée ou "complexe
activé" lequel se décomposerait pour donner les produits de réaction.
Henry Eyring (1901-1981)
Professeur à l'Université de
l'Utah (USA) apporta une contribution importante à la Théorie
du complexe activé..
Photo fournie gracieusement par l'Université
d'Utah, Salt Lake City, Utah 84112 USA.
Polanyi Michael
Professeur à l'Université de Manchester (Royaume Uni), co-publia
avec Eyring les travaux sur le complexe activé.
Photo fournie gracieusement par son fils,
Polanyi John Charles.
Polanyi John Charles (1929 - )
fils de Michael, Professeur à
l'Université de Toronto (Canada) reçut le Prix Nobel de
Chimie en 1986 pour ses travaux sur l'état de transition.
Cette théorie met donc l'accent sur
l'énergie du système au cours de la transformation.
L'origine de l'énergie d'activation n'entre pas en considération comme dans
la théorie des collisions.
De ce fait, la théorie du complexe activé peut être étendue aux réactions mono-moléculaires,
puisqu'il n'est pas nécessaire de faire appel à l'énergie du choc bi-moléculaire.
4.2.1 Présentation
simplifiée.
Considérons une réaction élémentaire
bimoléculaire entre réactifs A et B qui donne deux produits C et D.
La théorie du complexe activé pose
que les molécules A et B vont s'approcher l'une de l'autre mues par leur mouvement
propre, comme dans la théorie des collisions. L'énergie libre du système A
+ B augmente au fur et à mesure que les molécules s'approchent à cause des
interactions qui se manifestent de plus en plus fortement à courte distance
jusqu'au point où les molécules A et B ne peuvent plus être distinguées
dans leur individualité et qu'elles forment un complexe noté AB# d'énergie élevé.
Cecomplexe peut se décomposer en redonnant les réactifs A et B ou en donnant
les produits de la réaction.
La réaction est donc décrite par deux étapes
:
La formation du complexe activé par une réaction
réversible :
A +
B AB#
La transformation du complexe en produits
de la réaction :
AB# C
+ D
Ce qui constitue un mécanisme qui
peut être schématisé comme suit :
A
+ B
AB#
C + D
Si on construit un diagramme énergétique
en plaçant :
à gauche, le système
avant réaction (molécules A et B éloignées l'une de l'autre)
à droite, le système après réaction
(molécules C et D éloignées l'une de l'autre)
et au milieu le complexe activé,
on obtient :
La description précise de ce chemin
réactionnel et la caractérisation du complexe activé nécessite la description
simultanée de tous les atomes du système en cours de réaction. Cela ne peut
se faire que par les méthodes de la mécanique quantique. De ce fait, cette
description n'est possible que pour les systèmes les plus simples. Voir un
tel exemple paragraphe 4.2.4.
Pour les systèmes plus complexes,
on doit se contenter de décrire seulement les états initiaux et finaux et
d'essayer de caractériser le complexe activé. On doit même souvent simplifier
le système en ne retenant que les parties des molécules qui subissent des
transformation.
4.2.2 Vitesse
absolue de réaction.
L'objectif est d'établir une expression
de la vitesse de la réaction à partir de la connaissance de son mécanisme
réactionnel. Du fait que cette expression ne fait pas intervenir de facteur
empirique, on l'appelle souvent "vitesse absolue".
La vitesse de la réaction est égale
à la vitesse de formation des produits, donc à la vitesse de transformation
du complexe, soit : v = k# . CAB#
k# étant
assimilable au coefficient de vitesse de la réaction de décomposition du complexe
activé en produits.
Pour exprimer k# on doit faire appel des notions de mécanique
statistique qui dépassent le cadre de ce cours. On en donnera donc ici seulement
les résultats sans pouvoir en fournir les justifications. k# va être estimé en considérant que la décomposition
du complexe activé se fait par un mouvement similaire à une vibration interne
qui se produirait suivant la coordonnée de la réaction. On peut dès lors montrer
que la vitesse de la réaction s'exprime par
(1)
relation dans laquelle : kB est la constante de Boltzmann (kB
= 1,381.10-23 J.K-1), h la constante de Planck ( h = 6,626.10-34 J.s)
et T la température absolue de réaction. étant
le facteur de transmission.
représente la proportion de molécules activées, qui
franchissant le sommet de la barrière énergétique se transforment effectivement
pour donner les produits de réaction. Dans la plupart des cas , ce facteur
est égal à 1 (toutes les molécules activées qui franchissent le col donnent
les produits). Pour simplifier l'écriture, nous ne le ferons pas figurer dans
ce qui suit.
Pour plus de détails sur le facteur de transmission , cliquez
ici.
Pour évaluer CAB#on peut admettre qu'il s'établit un équilibre rapide entre les réactifs
et le complexe activé.
On peut donc écrire :
K#Cétant la constante d'équilibre relative aux concentrations entre les réactifs
et le complexe activé, d'où l'expression de la concentration de ce complexe
: CAB# = KC# . CA . CB
La vitesse de la réaction peut donc s'écrire (2) La loi de vitesse est donc du second ordre (ordre 1 par rapport à chaque
réactif) et son coefficient de vitesse k
est égal à .
On peut relier la constante K#C
à la constante thermodynamique d'équilibre K#entre les réactifs et le complexe activé.
En effet,
Po étant
la pression de référence (1bar), pA,
pB et pAB#
étant les pressions partielles de A, B et AB#.
Or pi = RT . ni / V = RT . Ci d'où (3)
La constante thermodynamique d'équilibre
K#est reliée à l'enthalpie libre standard
D#G°
de formation du complexe activé à partir des réactifs (voir le module de Thermodynamique).
Plus simplement, on appelle aussi D#G°
l'enthalpie libre d'activation.
En effet, la considérant l'équilibre entre les réactifs et le complexe activé
: A + B AB#
La constante thermodynamique K# de cet
équilibre est reliée à D#G°
par D#G° = - RT . ln K#
Le coefficient de vitesse s'écrit
donc (4)
D'après la définition de l'enthalpie
libre G = H - T.S , on peut décomposer
l'enthalpie d'activation D#G°
en un terme enthalpique
et un terme entropique : D#G° = D#H° -
T.D#S°
D'où l'expression du coefficient
de vitesse (5)
Voyons comment cette expression
peut être reliée à l'équation d'Arrhenius :
en en prenant le log et en dérivant on obtient : (6)
De même, en prenant le logarithme
de la relation (5) et en dérivant on trouve :
(7)
En identifiant les relations (6)
et (7) il vient : Ea = 2.R.T + D#H°
(8)
d'où : D#H° = Ea
- 2.R.T
Et l'expression du coefficient de
vitesse de la réaction :
(9)
D'où l'expression théorique du facteur
de fréquence A de la relation d'Arrhenius :
(10)
On voit donc que l'énergie d'activation
d'Arrhenius, qui est d'origine empirique, est reliée à l'enthalpie standard
de formation du complexe activé à partir des réactifsD#H°
(relation 8) et que l'expression théorique du facteur de
fréquence fait intervenir l'entropie standard de formation du complexe activé
D#S°
(relation 10)
C'est par ces deux grandeurs que la théorie du complexe activé prend en compte
la nature chimique des réactifs et de la façon dont ils réagissent, chose
que ne pouvait pas faire la théorie des collisions sauf en introduisant à
posteriori le facteur stérique.
Conclusion
: Dans la présentation simplifiée de la théorie du complexe activée
qui a été donnée ici, nous nous sommes restreint au cas des réactions bimoléculaires
en phase gazeuse dont l'énergie d'activation est d'origine thermique. La théorie
peut être adaptée au cas des réactions mono-moléculaires activées photochimiquement
et des réactions en solution.
Cette théorie permet le calcul du coefficient de vitesse d'une réaction sans
introduire de facteur empirique.
Dans les cas les plus simples, l'accord entre les valeurs théoriques et expérimentales
est acceptable. La difficulté inhérente à la méthode réside dans la détermination
du chemin réactionnel et de la structure précise du complexe activé. Dans
ces cas les plus simples, on peut construire la surface d'énergie potentielle
pour caractériser ce chemin réactionnel et identifier le complexe activé.
Cette approche est illustrée dans le paragraphe suivant.
Dans les cas plus complexes, une analyse exhaustive est impossible et on doit
modéliser l'approche des réactifs et leur mode d'interaction avec la plus
grande prudence.
4.2.3 Surfaces
d'énergie potentielle.
Pour illustrer plus précisément
la notion de chemin réactionnel et d'état de transition, nous construirons
la surface d'énergie potentielle dans le cas le plus simple où un atome d'hydrogène
vient réagir avec une molécule de dihydrogène pour remplacer un de ces atomes.
Pour pouvoir distinguer les trois
atomes participant à la réaction, nous les indicerons par les lettres A, B,
C.
La réaction considérée s'écrit :
HA + HB-HC = HA-HB + HC
Ou encore plus simplement, pour
alléger la notation :
A + BC = AB + C
L'atome A peut approcher la molécule
BC selon toutes les directions. Les calculs d'énergie montrent que l'approche
de l'atome A le long de l'axe moléculaire B-C est la direction qui requiert
le moins d'énergie. Pour simplifier nous n'envisagerons que cette direction
d'approche.
Examinons les modes de vibrations
dans l'état de transition :
En résumé :
Dans le cas présent, nous avons
simplifié le problème en ne considérant que les positions dans lesquelles
les trois atomes restent alignés.
Ceci est justifié car on peut montrer que lorsque l'atome d'hydrogène arrive
ou part selon une direction différente de l'axe de la molécule de dihydrogène,
la surface d'énergie potentielle est déformée et se trouve à une énergie supérieure
à celle obtenue lorsque les trois atomes restent alignés. On n'a donc à
considérer que les modes de vibration d'élongation (ce qui ne veut pas dire
que les vibrations de déformation angulaire n'existent pas, mais qu'ils correspondent
à des énergies plus élevées).
Dans la surface d'énergie potentielle
les deux modes de vibration correspondent à des déplacements du point représentatif
du système selon la bissectrice pour la vibration symétrique et sa perpendiculaire
pour la vibration antisymétrique. Il est facile de voir que la vibration symétrique
n'a pas d'influence sur la réaction considérée, tandis que la vibration antisymétrique
s'effectue en quelque sorte le long du chemin réactionnel. Lorsque l'amplitude
de cette vibration augmente, elle conduit à la rupture de l'état de transition
et à la formation des produits de réaction ou des réactifs selon que l'élongation
positive concerne la liaison BC ou AB.
C'est donc la vibration antisymétrique qui permet le passage par l'état de
transition.
Notes sur le parcours sur la
surface d'énergie potentielle.
Le chemin réactionnel a été défini
comme la suite d'états du système qui permet de passer de l'état initial (réactifs)
à l'état final (produits) avec l'apport minimum d'énergie pour franchir la
barrière d'énergie potentielle. Comme son nom l'indique, la surface d'énergie
potentielle représente seulement l'énergie potentielle à laquelle il faut
ajouter l'énergie cinétique (translation, rotation, vibration) de tous les
constituants du système pour avoir son énergie totale. Si on considère le
système dans son état initial (A + BC), la molécule BC a un mouvement
de vibration qui lui confère une énergie cinétique de vibration. En tenant
compte de ce mouvement, le point représentatif du système n'est plus fixe
mais se déplace de part et d'autre de la position d'équilibre.
Si maintenant on considère l'approche
des deux réactifs (A et BC) le long de l'axe BC, on doit ajouter un terme
d'énergie cinétique qui place le point représentatif du système à une
certaine hauteur par rapport à la surface d'énergie potentielle. Au fur et
à mesure que les réactifs s'approchent l'un de l'autre, la projection du point
représentatif du système sur le plan va se déplacer comme sur l'animation.
Au fur et à mesure que le système
"remonte" vers le col, son énergie potentielle augmente et il perd de l'énergie
cinétique (ou son énergie cinétique diminue d'autant). Si l'énergie cinétique
du système est suffisante, le système va pouvoir atteindre l'état de transition
avec une énergie de vibration antisymétrique suffisante pour le franchir et
donner les produits de réactions. Lors de la séparation des produits, le système
regagne de l'énergie cinétique en perdant de l'énergie potentielle.
Si l'énergie cinétique du système
n'est pas suffisante, le système ne va pas pouvoir atteindre l'état de transition.
Il va donc "revenir" en arrière et les réactifs vont se séparer sans avoir
réagi.
Généralisation.
Dans la présentation ci-dessus,
nous avons étudié le cas le plus simple qui soit : trois atomes identiques
(d'hydrogène de surcroît), approchant l'un de l'autre selon une direction
privilégiée. Cela nous a permis de définir l'énergie potentielle du système
comme étant fonction de deux paramètres seulement (rAB et rBC)
donc de représenter la surface d'énergie potentielle dans un espace à trois
dimensions.
Il est alors possible de développer
les calculs de mécanique quantique et de thermodynamique statistique dans
le cadre de la théorie du complexe activé et d'obtenir des valeurs des coefficients
de vitesse avec une bonne précision. Un premier niveau de complexité apparaît
dès que l'on s'intéresse à trois atomes qui ne sont pas identiques. La surface
d'énergie potentielle n'est plus symétrique mais les calculs restent possibles
et les résultats dépendent de la précision de ces calculs.
Dès que l'on a à faire a des systèmes
plus complexes comportant plus de trois atomes, il devient impossible de représenter
la surface d'énergie potentielle. Mais les concepts développés ci-dessus restent
valables. On doit donc imaginer le chemin réactionnel et le valider en effectuant
des calculs ponctuels en une série de points caractéristiques de ce chemin.
Un des points délicats réside dans la caractérisation de l'état de transition
et de ses modes de vibration. En effet, dans l'exemple développé ci-dessus,
la nature de la vibration qui est responsable du passage par l'état de transition
est facile à identifier. Dans le cas de réactions faisant intervenir des réactifs
plus complexes, la vibration correspondant au passage par l'état de transition
peut faire intervenir de nombreux atomes et ne peut pas toujours être facilement
associée à une liaison particulière. Cependant, le concept de vibration de
l'état de transition s'effectuant " selon le chemin réactionnel " demeure.
Chapitre 4 Bases de la théorie...
Nature des processus
élementaires
et des intermédiaires.



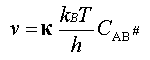 (1)
(1)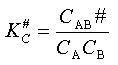
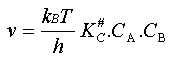 (2)
(2)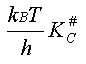 .
.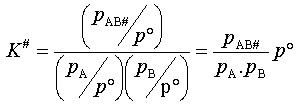
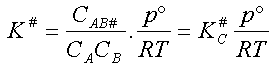 (3)
(3)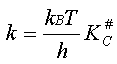 s'écrit
donc
s'écrit
donc 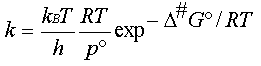 (4)
(4)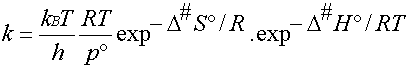 (5)
(5)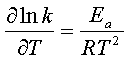 (6)
(6) 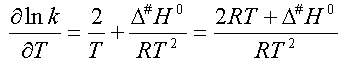 (7)
(7) 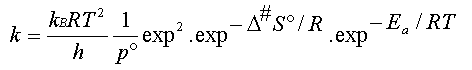 (9)
(9) 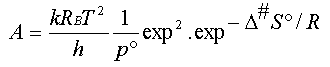 (10)
(10)