La vitesse d'une réaction est définie par la variation des quantités de
réactifs ou de produits observée au cours du temps.
Il s'agit donc d'une grandeur macroscopique
qui s'applique à un grand ensemble de molécules.
Il en est de même des concepts qui en dérivent tels que la loi de vitesse et
l'ordre de réaction. Du point de vue microscopique, on essaie de se représenter
la réaction au niveau des molécules individuelles. La transformation chimique
se traduit par une redistribution des électrons et / ou d'atomes qui s'accompagne
de la rupture de certaines liaisons et l'établissement d'autres liaisons
chimiques.
La théorie de la réaction chimique essaie d'établir le lien entre les observations
effectuées au niveau macroscopique et la manière dont se passe la réaction au
niveau moléculaire.
Pour développer cette théorie, on considère séparément les réactions s'effectuant
en une seule étape. De telles réactions qui ne peuvent pas être décomposées
en étapes plus simples sont appelées réactions élémentaires
ou processus élémentaires.
4.1.1 Chocs
bimoléculaires.
Nous considèrerons tout d'abord les réactions bimoléculaires en phase
gazeuse.
Une réaction bimoléculaire, A + B ne peut se produire que lorsque les
deux entités chimiques A et B entrent en contact.
La vitesse de la transformation est donc proportionnelle au nombre de chocs
bimoléculaires entre A et B par unité de temps (fréquence des
chocs).
Il est assez intuitif de comprendre que la fréquence de ces chocs est elle-même
proportionnelle à la concentration CA et CB
de chacune des entités.
Fréquence des collisions µCA.CB
Le symbole µ
est utilisé pour noter la proportionnalité.
Notez que cette relation de proportionnalité avec les concentrations
peut être établie par une démonstration géométrique en estimant la fréquence
des chocs à partir de la vitesse moyenne des molécules dans la phase gazeuse.
Or, cette vitesse moyenne dépend de la température. Plus les molécules se déplacent
vite, plus grande sera la fréquence des chocs.
En tenant compte de la distribution des vitesses des molécules dans le gaz,
on aboutit à une expression qui fait intervenir la masse et la taille des molécules,
ainsi que la température par un facteur en .
Une telle démonstration dépasse le niveau de cette présentation.
Vous pouvez voir une illustration de l'influence
de la masse moléculaire sur la distribution des vitesses des molécules dans
un gaz.
Vous pouvez voir une illustration de l'influence de la
température sur la distribution des vitesses des molécules dans un gaz.
Dans le modèle simple utilisé ci-dessus pour calculer la vitesse des molécules
dans le gaz, on considère les molécules comme des masses ponctuelles n'ayant
aucune interactions entre elles. On peut affiner ce modèle en assimilant les
molécules à des sphères et définir ainsi une section de choc qui tient compte
de la taille des molécules.
A partir de ce modèle on peut estimer la fréquence des chocs bimoléculaires.
Le résultat auquel on aboutit fait intervenir trois facteurs :
Un facteur en avec
m la masse réduite des molécules A et B qui entrent en collision
:
mA et mB étant respectivement les masses molaires de A et B
Un facteur en s
(appelé section de choc) qui dépend des rayons des sphères représentant
les molécules A et B.
Un facteur en qui
représente l'influence de la température sur la fréquence des collisions.
Pour avoir l'expression complète de
la fréquence des chocs bimoléculaires, cliquez
ici
Le modèle simple selon lequel tous les chocs entre
molécules donneraient lieu à une réaction n'est pas recevable pour trois
raisons :
Les vitesses de réactions calculées directement
à partir de la fréquence des chocs bimoléculaires sont beaucoup plus
grandes que les valeurs observées expérimentalement. La divergence peut
atteindre 10 12 à 10 20 !
L'influence
de la température sur la vitesse de réaction suit généralement l'équation
d'Arhenius,
c'est à dire fait intervenir un terme en e -E / kT
et non pas seulement un terme en
.
On constate que les ordres de grandeurs
des vitesses de réactions varient dans des proportions énormes (de 1
à 10 5). Autrement dit, il y a des réactions extrêmement
rapides et d'autres extrêmement lentes. L'échelle de ces variations
est beaucoup plus grande que ne laisse prévoir l'influence de la
taille et de la masse des molécules (de 1 à 10 2).
On doit donc considérer que tous les chocs
bimoléculaires ne sont pas efficaces.
Deux facteurs sont à prendre en compte :
Premièrement, il faut que le choc entre les
deux molécules se fasse avec une énergie suffisante pour que les liaisons
puissent se rompre.
Deuxièmement, il faut que les molécules se présentent
dans une orientation favorable pour que la réaction puisse avoir lieu.
4.1.2 Energie d'activation.
L'énergie cinétique des molécules dépend de leur
masse et de leur vitesse .
Or, dans le gaz, toutes les molécules n'ont pas la même vitesse. La distribution
des vitesses des molécules dans le gaz a été étudiée au paragraphe précédent.
De façon similaire, on peut étudier la distribution de l'énergie cinétique des
molécules.
Celle-ci s'établit à partir de l'expression de l'énergie cinétique et
de la fonction de distribution des vitesses.
Vous pouvez voir ci-dessous une illustration de
la distribution de l'énergie cinétique des molécules.
La proportion de molécules ayant une
énergie suffisante est donc proportionnelle au facteur de Boltzmann .
Il en résulte que la vitesse v d'une réaction bimoléculaire
devrait s'exprimer par une relation de la forme :
v
µ
[Fréquence des collisions].[Proportion de molécules ayant une énergie suffisante]
Le symbole µ
est utilisé pour noter la proportionnalité.
Pour deux molécules A et B différentes
de diamètres respectifs dA et dB et
de masses mA et mB, le facteur [Fréquence
des collisions] (noté ZAB) est lui même proportionnel
à avec
s la section de choc qui se calcule
par s = p.d2
avec d = ½ (dA + dB)
m la masse réduite qui se calcule
par ,
et N est la constante d'Avogadro.
Le facteur [Proportion de molécules ayant une
énergie suffisante] est proportionnel à .
On obtient :
En comparant cette relation avec celle
de la loi de vitesse du second ordre v = k.CA.CB,
on en déduit que, dans le cadre de cette théorie, le coefficient de vitesse
k prend la forme :
En première approximation, cette relation
est compatible avec l'équation empirique d'Arrhenius sous
réserve que le facteur exponentiel l'emporte sur la variation en qui
interviendrait dans le facteur pré-exponentiel A.
Pour lire une note sur cette approximation, cliquez
ici.
4.1.3 Facteur stérique.
Dans ce qui précède, on a assimilé les molécules
a de simples sphères de diamètres dA et dB.
En fait, ceci n'est vrai que pour des atomes ou des ions monoatomiques.
Dans tous les autres cas, il faudrait tenir compte
de leur forme réelle pour pouvoir calculer avec une meilleure approximation
la section de choc s entre les deux molécules,
Cette section de choc représente en quelque sorte la surface effective moyenne
de rencontre des molécules.
En pratique, même si on estime convenablement la
section de choc entre deux molécules en tenant compte de leur forme, ce qu'il
n'est possible de faire que dans des cas simples, les coefficients de vitesse
estimés théoriquement par la relation ont
généralement beaucoup plus grands que les valeurs déterminées expérimentalement.
Cela s'interprète en considérant que, même s'ils ont une énergie suffisante,
tous les chocs ne conduisent pas nécessairement la transformation. On dit que
tous les chocs ne sont pas efficaces.
Pour tenir compte du fait que seuls
les chocs se produisant suivant la bonne direction et avec la bonne orientation
peuvent conduire à la transformation chimique, on définit la section de choc
efficace s* = P.s
, P étant un facteur de proportionnalité plus petit que 1. Ce facteur est
parfois appelé facteur de probabilité ou facteur d'Hinshelwood du nom du physico-chimiste
anglais Sir Cyril Norman d'Hinshelwood qui l'a introduit dans les années 1935,
prix Nobel 1956.
Cyril Norman Hinshelwood est un physico-chimiste
britannique né à Londres le 19 juin 1897 et mort à Londres en 1967.
Il
fit ses études à l'université d'Oxford et y obtint son doctorat avant
d'y être nommé professeur en 1937. Hinshelwood reçut en 1956 le prix Nobel
de chimie (qu'il partagea avec Nikolay Nikolaevich Semenov) pour ses études
cinétiques des réactions en phase gazeuse.
Il
fut anobli en 1948 et, président de la Royal Society de 1955 à 1960.
L'expression théorique du coefficient de vitesse
s'écrit donc :
Expression que l'on identifie facilement avec la loi empirique d'Arrhenius
En pratique, on détermine le facteur de proportionnalité
P en comparant le terme théorique avec
la valeur du facteur préexponentiel A de la loi d'Arrhenius déterminée
expérimentalement.
L'étude expérimentale de la réaction aux environs
de 600 K considérée ici comme exemple :
donne les valeurs suivantes : A
= 1,2.106 L.mol-1.s-1 et Ea
= 180 kJ.mol-1 .
La valeur théorique du terme est
7,3.1011 L mol-1.s-1> d'où P = 1,7.10-6
. Cette valeur est très faible.
Toutes les réactions n'ont pas des valeurs
aussi faibles du facteur stérique.
Par exemple, la réaction 2NO22NO + O2
a un facteur stérique de l'ordre de 6.10-2
Il existe même des réactions pour
lesquelles le facteur stérique est proche de 1,
c'est le cas par exemple de C2H5Br + HO-C2H5OH
+ Br-.
Pour voir laThéorie
des processus pseudo monomoléculaires de Lindemann, cliquez
ici.
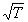 .
Une telle démonstration dépasse le niveau de cette présentation.
.
Une telle démonstration dépasse le niveau de cette présentation.
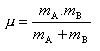
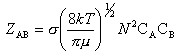 avec
avec
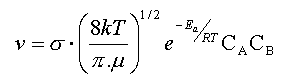
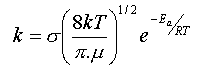
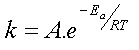 sous
réserve que le facteur exponentiel l'emporte sur la variation en
sous
réserve que le facteur exponentiel l'emporte sur la variation en 
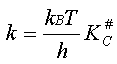
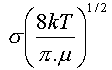 avec
la valeur du facteur préexponentiel A de la loi d'Arrhenius déterminée
expérimentalement.
avec
la valeur du facteur préexponentiel A de la loi d'Arrhenius déterminée
expérimentalement.